그동안 단백질을 읽어내는 일은 DNA를 읽는 것보다 훨씬, 아주 훨씬 어려웠습니다. DNA는 단 4종류의 염기로 구성되어 시퀀싱 기술이 비약적으로 발전했지만, 단백질은 20종류의 아미노산으로 이루어진 데다 구조까지 복잡하기 때문입니다. 하지만 2026년 3월, 전 세계 과학계를 뒤흔든 혁명적인 생명정보학(Bioinformatics) 연구들이 발표되면서 단백질과 유전체의 비밀이 속속들이 밝혀지고 있습니다.
마치 영화 <스타트렉>의 ‘트라이코더’처럼 샘플만 넣으면 모든 생체 정보를 읽어내는 시대가 코앞으로 다가온 것이죠. [1]
단백질 시퀀싱의 대혁명: ‘역번역’ 기술의 탄생
그동안 단백질 분석의 표준은 ‘질량 분석법(Mass Spectrometry)’이었습니다. 하지만 이 방식은 수십억 개의 분자가 필요하고, 아주 적은 양으로 존재하는 희귀 단백질은 찾아내기 어렵다는 단점이 있었죠.
2026년 3월 18일, 스탠포드 대학교 연구팀은 Nature Biotechnology를 통해 이 거대한 장벽을 무너뜨리는 ‘단일 분자 펩타이드 시퀀싱’ 기술을 발표했습니다. [1]
단백질을 DNA 바코드로 변환하여 읽다
연구팀이 개발한 기술의 핵심은 역발상입니다. 단백질을 직접 읽는 대신, 단백질의 아미노산 서열 정보를 DNA 바코드로 ‘기록’한 뒤, 이미 전 세계적으로 보급된 저렴하고 빠른 DNA 시퀀서(NGS)로 읽어내는 방식입니다.
이를 연구진은 ‘역번역(Reverse Translation)’이라 명명했습니다. [1]
이 과정은 다음과 같은 3단계로 진행됩니다.
- 인코딩(Encoding): 자석 비드 위에 단백질 조각(펩타이드)을 고정하고, 각 분자마다 고유한 DNA 바코드를 부여합니다.
- 순차적 기록: 단백질의 끝에서부터 아미노산을 하나씩 떼어내면서(에드만 분해법 변형), 해당 아미노산이 무엇인지 알려주는 DNA 리포터를 생성합니다.
- 라이브러리 구축 및 시퀀싱: 생성된 DNA 리포터들을 모아 한 번에 시퀀싱하면, 원래 단백질의 어떤 위치에 어떤 아미노산이 있었는지 디지털 정보로 완벽하게 복원할 수 있습니다.
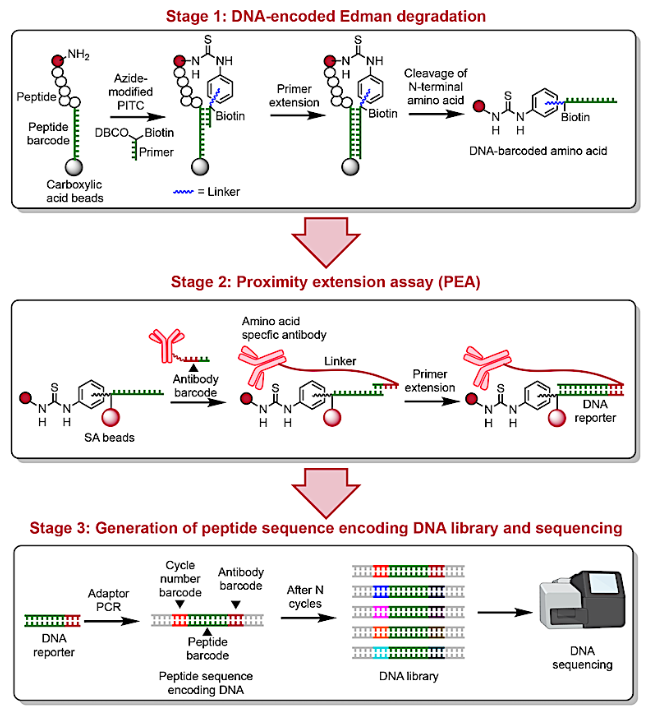
1단계에서 펩타이드는 자성 비드에 고정된 펩타이드 바코드 DNA 서열과 접합됩니다.
아지드기로 변형된 PITC 분자를 사용하여 변형된 에드만 분해 반응을 수행함으로써, DBCO로 변형되고 비오틴화된 프라이머와 클릭 커플링을 통해 결합하고, 이 프라이머는 펩타이드 바코드 서열에 직접 결합합니다.
이 프라이머는 N말단 아미노산 절단 전에 펩타이드 바코드를 기록하도록 확장됩니다.
2단계에서 절단 후 방출된 DNA 바코드 아미노산은 비오틴기를 통해 스트렙타비딘(SA) 비드에 의해 고정됩니다.
이렇게 고정된 아미노산은 DNA 바코드 아미노산 특이적 항체에 의해 인식되고, 근접 확장 분석(PEA)을 통해 DNA 리포터로 변환됩니다.
- 고정된 아미노산을 인식하는 특이적 항체에는 해당 아미노산의 종류를 나타내는 DNA 서열(DNA Barcode)이 꼬리표처럼 붙어 있습니다.
- 항체가 아미노산에 결합하면, 항체에 달린 DNA 바코드 정보가 근접 확장 분석(PEA) 과정을 통해 DNA 리포터(DNA Reporter) 서열로 변환됩니다. 이 리포터에는 결합한 아미노산의 정체 정보가 담깁니다.
3단계에서 각 주기에서 생성된 DNA 리포터는 어댑터 PCR 과정에서 주기 번호 바코드로 바코딩되어, 각 아미노산의 종류, 위치 및 유래 펩타이드를 인코딩하는 DNA 서열을 생성합니다.
이 과정을 통해 생성된 최종 DNA 라이브러리는 단일 실행으로 시퀀싱할 수 있으므로 샘플로부터 펩타이드 서열을 재구성할 수 있습니다.
💡 개념 쏙쏙: 에드만 분해법(Edman Degradation)
에드만 분해법은 단백질의 N-말단(시작점)부터 아미노산을 하나씩 차례대로 잘라내어 그 종류를 확인하는 고전적인 화학 반응입니다.
1950년대에 개발된 이 기술은 그동안 너무 느리고 샘플 소모가 많아 외면받았지만, 2026년의 연구진은 여기에 ‘DNA 바코딩’을 접목하여 단일 분자 수준까지 감도를 끌어올린 최첨단 도구로 부활시켰습니다. [1, 5]
왜 이 기술이 ‘게임 체인저’일까요?
가장 놀라운 점은 감도입니다. 기존 질량 분석법보다 약 1,000배나 더 민감하여, 단 하나의 세포 속에 들어있는 극미량의 단백질까지 잡아낼 수 있습니다. [1]
| 비교 항목 | 기존 질량 분석법(MS) | 새로운 역번역 기술 |
|---|---|---|
| 필요 샘플 양 | 10억~100억 개의 분자 | 단일 분자 수준 (Single-molecule) |
| 검출 효율 | 약 1/1,000 수준 | 이론상 모든 분자 확인 가능 |
| 분석 장비 | 고가의 전용 질량 분석기 | 기존 DNA 시퀀싱 플랫폼 활용 |
| 주요 활용 | 벌크 샘플 분석 | 단일 세포 수준의 단백질체 분석 |
이 기술 덕분에 과학자들은 왜 똑같은 암세포인데도 어떤 녀석은 면역 항암제(CAR-T)에 반응하고 어떤 녀석은 살아남는지, 그 미세한 단백질 수준의 차이를 나노미터 해상도로 들여다볼 수 있게 되었습니다.
현재 이 기술은 “샘플만 넣으면 결과가 나오는” 버튼식 기기로 상용화가 진행 중입니다. [1]
인공지능이 그리는 생명의 지도: AlphaGenome과 Bio-Copilot
2026년 3월, 생명정보학 분야의 인공지능(AI)은 단순한 보조 도구를 넘어 가설을 스스로 세우고 검증하는 ‘동료 과학자’의 단계로 진입했습니다. 특히 구글 딥마인드와 주요 대학 연구소들의 발표는 충격적이기까지 합니다. [6, 7, 8]
게놈의 숨겨진 스위치를 찾는 AlphaGenome
구글 딥마인드가 공개한 AlphaGenome은 무려 100만 염기쌍(1Mb)의 긴 DNA 서열을 한꺼번에 분석하는 딥러닝 모델입니다.
인간 게놈의 대부분을 차지하는 ‘비부호화 영역(Non-coding region)’은 그동안 쓰레기 DNA로 취급받기도 했지만, 실제로는 유전자의 작동을 조절하는 복잡한 스위치들이 가득합니다. [7]
AlphaGenome은 DNA 서열만 보고 다음과 같은 11가지 요소를 예측합니다.
- 유전자 발현: 특정 조직에서 유전자가 얼마나 켜질 것인가?
- RNA 스플라이싱: 유전 정보가 어떻게 편집되어 단백질로 변할 것인가?
- 염색질 접근성: DNA의 어느 부위가 열려 있어 조절 인자가 붙을 수 있는가?
- 3차원 구조: 멀리 떨어진 DNA 부위들이 공간적으로 어떻게 만나 조절 신호를 주고받는가? [7, 8]
이 모델은 싱글 베이스(Single-base) 해상도를 지원하기 때문에, 단 하나의 염기만 바뀌어도 질병 위험이 어떻게 변하는지 정확히 예측할 수 있어 정밀 의료의 핵심 자산이 되고 있습니다. [7, 8]
가설 없는 탐사: Bio-Copilot 시스템
광저우 국립 연구소와 주요 대학들이 협력하여 발표한 Bio-Copilot은 거대언어모델(LLM)을 기반으로 한 데이터 지능형 시스템입니다.
과거의 연구 방식이 “A 유전자가 암을 일으킬 것이다”라는 가설을 세우고 실험하는 방식이었다면, Bio-Copilot은 방대한 오믹스(Omics) 데이터를 스스로 탐사하여 우리가 미처 생각지 못한 새로운 생물학적 패턴을 찾아냅니다. [6]
Bio-Copilot은 실제로 대규모 인간 폐 세포 아틀라스(Atlas)를 구축하면서, 세포의 상태가 불연속적인 단계가 아니라 연속적인 변화 과정에 있음을 정교하게 주석(Annotation)하는 데 성공했습니다.
이는 AI가 인간 전문가의 지식과 결합했을 때 생물학적 발견의 속도를 얼마나 가속할 수 있는지를 보여주는 완벽한 사례입니다. [6, 9]
💡 개념 쏙쏙: 오믹스(Omics)란?
오믹스는 ‘-ome(전체)’과 ‘-ics(학문)’의 합성어로, 개별 유전자나 단백질 하나가 아니라 세포 내에 존재하는 해당 구성 요소 전체를 한꺼번에 연구하는 학문을 말합니다.
게놈(유전체), 전사체(RNA 전체), 단백질체(단백질 전체) 등이 대표적입니다. 2026년의 생명정보학은 이 여러 층위의 데이터를 하나로 통합하는 ‘멀티 옴닉스’가 주류입니다. [3, 10, 11]
희귀 질환의 유전적 암호: GOLGA8A 반복 서열
유전학 연구에서 가장 풀기 어려운 숙제 중 하나가 바로 ‘가족력 없는 환자(산발성 환자)’의 발병 원인을 찾는 것이었습니다.
2026년 3월 Nature Genetics에는 조기 발병형 치매 중 하나인 전두측엽 치매(FTD)의 새로운 원인 유전자가 발표되었습니다. [12, 13, 14]
‘보이지 않는 변이’를 찾아낸 롱리드 시퀀싱
연구진은 벨기에의 안트베르펜 대학교와 VIB 연구소 팀으로 구성되었으며, 수천 명의 대규모 코호트 데이터를 분석했습니다.
그들은 일반적인 분석 기술로는 절대 볼 수 없었던 GOLGA8A 유전자의 인트론(비부호화 부위) 내 ‘CT’ 두 글자의 무한 반복 확장 현상을 발견했습니다. [12, 14, 15]
| 분석 항목 | 상세 연구 결과 |
|---|---|
| 관련 질환 | 산발성 전두측엽 치매 (aFTLD-U 서브타입) |
| 원인 유전자 | GOLGA8A (15번 염색체 위치) |
| 변이 특징 | CT-다이머 탄뎀 반복 서열의 비정상적 확장 |
| 발견 의미 | 해당 환자군의 약 60%에서 발견되는 매우 강력한 위험 인자 |
| 기술적 성과 | 롱리드 시퀀싱을 통한 복잡한 반복 구간의 완벽한 해독 |
그동안 이 변이가 발견되지 않았던 이유는 우리 게놈에 GOLGA8A와 아주 유사한 복사본들이 수십 개나 존재하기 때문입니다.
기존의 짧게 읽는 방식는 어디가 원본이고 어디가 변이인지 구별할 수 없었죠. 하지만 2026년의 성숙한 롱리드 기술은 이 복잡한 퍼즐을 한 번에 풀어냈습니다. [13, 14, 15]
공간 생물학: 조직 사진 한 장으로 유전체 지도를 그리다
과거에는 조직 내 유전자의 위치를 알기 위해 수억 원대의 장비와 수개월의 시간이 필요했습니다.
하지만 2026년 3월, 이 과정을 극적으로 단순화한 연구들이 발표되었습니다.
그 중심에는 인공지능과 공간 전사체학(Spatial Transcriptomics)의 융합이 있습니다. [16, 17, 18]
SHEST 모델
삼성서울병원과 성균관대학교 연구팀이 발표한 SHEST는 병원에서 흔히 찍는 일반적인 현미경 사진(H&E 염색 이미지)을 입력하면, 그 조직 내 각 세포가 어떤 종류인지 맞추고, 그 위치에서 어떤 유전자들이 발현되고 있는지까지 예측해 줍니다. [16]
이 기술의 핵심 메커니즘은 다음과 같습니다.
- 다중 작업 학습: 세포의 모양(형태학)과 유전자 발현 데이터를 동시에 학습하여, 시각적 패턴과 분자적 특징 사이의 상관관계를 완벽히 파악합니다. [16]
- 이웃 정보 활용: 개별 세포 하나만 보는 것이 아니라, 주변 세포들과의 관계를 분석하여 모호한 신호를 필터링합니다. [16]
- 고해상도 복원: 폐암 데이터 분석 결과, 종양 세포와 면역 세포의 경계선을 90% 이상의 정확도로 찾아냈으며, 종양 미세환경 내의 복잡한 세포 간 상호작용 지도를 그려냈습니다. [16]
이러한 기술은 의료 현장에서 즉각적인 변화를 불러옵니다.
값비싼 전사체 분석을 할 여력이 없는 중소 병원에서도, AI 모델만 있다면 일반 조직 사진만으로 정밀 의료 서비스를 제공할 수 있게 된 것이죠.
이는 암 환자의 개인별 맞춤형 치료 전략을 세우는 데 결정적인 역할을 합니다. [2, 16, 19]
💡 개념 쏙쏙: 공간 전사체학(Spatial Transcriptomics)
기존의 RNA 시퀀싱이 과일을 모두 갈아서 주스로 만든 뒤 성분을 분석하는 것이라면, 공간 전사체학은 과일 샐러드에서 어떤 과일 조각이 어느 위치에 있는지 그대로 둔 채 유전 성분을 분석하는 기술입니다.
암세포가 주변의 정상 세포나 면역 세포와 어떻게 대화하며 세력을 확장하는지 연구할 때 필수적입니다. [16, 17, 20]
면역 시스템의 암호를 해독하는 기하학적 딥러닝
우리 몸의 파수꾼인 T세포가 적(바이러스, 암세포)을 어떻게 인식하는지 알아내는 것은 백신 개발의 성배와도 같습니다.
2026년 3월 Briefings in Bioinformatics는 이 복잡한 상호작용을 수학적으로 풀어낸 연구들을 게재했습니다. [3, 21]
면역학적 지문을 읽다
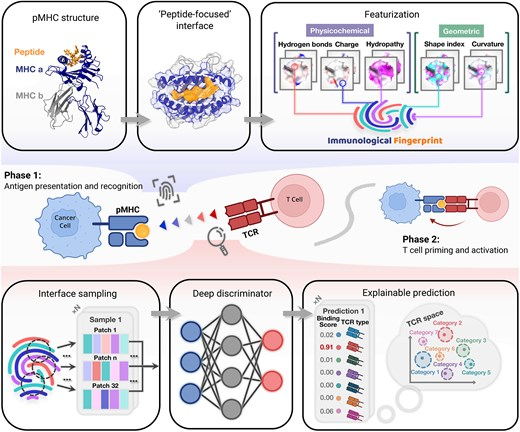
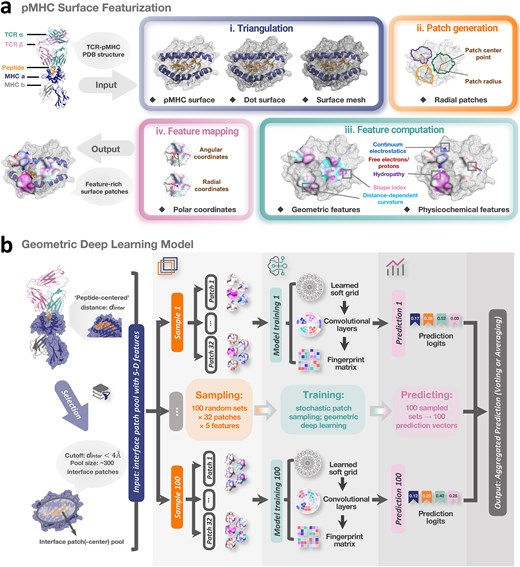
T세포 수용체(TCR)와 항원이 결합하는 과정은 마치 복잡한 열쇠와 자물쇠의 관계와 같습니다.
연구진은 기하학적 딥러닝(Geometric Deep Learning)을 도입하여, 이들의 3차원 입체 구조를 ‘면역학적 지문’으로 변환했습니다. [3, 21]
이 기술의 놀라운 성과는 다음과 같습니다.
- 미지의 변이 예측: 과거에 학습하지 않았던 새로운 바이러스 변이(OOD, Out-of-distribution)에 대해서도 T세포가 어떻게 반응할지 높은 확률로 예측합니다. [21, 22]
- 개인 맞춤형 암 백신: 환자 본인의 암세포가 가진 고유한 돌연변이(Neoantigen)를 타겟팅하는 최적의 T세포를 디자인할 수 있습니다. [3, 21]
- 약물 재창출: 이미 승인된 약물들 중 면역 세포의 활동을 돕는 보조제를 AI가 순식간에 골라냅니다. [10]
바이러스의 생존 전략: 원형 RNA ‘circHIV’
반면 바이러스도 만만치 않습니다. 예일 대학교 연구팀은 HIV-1 바이러스가 자신의 게놈을 원형으로 말아 ‘원형 RNA(circRNA)’를 만든다는 사실을 발견했습니다.
circHIV라 불리는 이 분자는 바이러스의 증식을 돕는 엔진 역할을 하며, 숙주 세포의 감시망을 피합니다. [23]
연구팀은 정교한 생명정보학 파이프라인을 가동하여 HIV 게놈 전체에서 9개의 원형 RNA 후보를 찾아냈고, 그중 실제 감염 상태에서 활발히 작동하는 녀석들을 검증했습니다.
이 발견은 바이러스의 아킬레스건을 공략하는 새로운 항바이러스제 개발의 단초가 되었습니다. [23]
바이오 빅데이터: 알고리즘과 인프라의 혁신
아무리 훌륭한 이론이 있어도, 수 테라바이트(TB)의 데이터를 분석하는 데 한 달이 걸린다면 실용성이 떨어지겠죠. 2026년 3월에는 분석의 ‘속도’와 ‘효율’을 극한으로 끌어올린 소프트웨어 도구들이 대거 등장했습니다. [2]
GPU 기반의 유전체 연구: SAIGE-GPU
우리가 게임할 때 주로 쓰는 그래픽 카드(GPU)가 이제 생명과학 연구의 심장이 되었습니다.
SAIGE-GPU는 수천 명 이상의 전장 유전체 연관 분석(GWAS)을 수행할 때, 기존 CPU 방식보다 수십 배 빠른 속도를 보여줍니다. [2]
또한, 클라우드 환경 최적화를 통해 분석 비용을 극적으로 낮춘 사례도 보고되었습니다.
- r7a.12xlarge 인스턴스: 기존 대비 분석 시간을 33% 단축하고 비용을 37% 절감했습니다. [8]
- r8i.8xlarge 인스턴스: 기준 모델과 유사한 성능을 내면서도 비용은 무려 61%나 저렴하게 처리할 수 있음을 확인했습니다. [8]
마이크로바이옴의 구글: Kun-peng 분류기
우리 몸속 미생물 생태계(마이크로바이옴) 데이터는 그 양이 방대하여 정체가 무엇인지 분류하는 것조차 고된 일이었습니다.
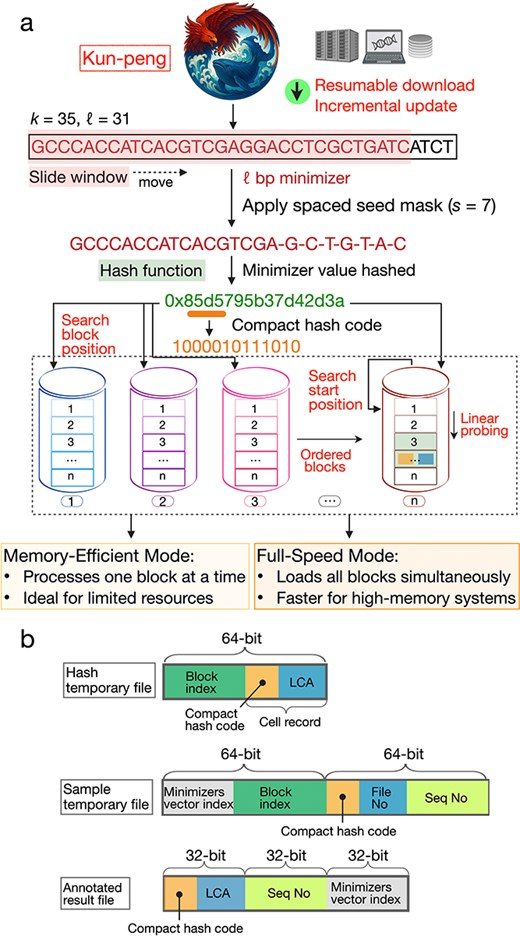
새롭게 개발된 Kun-peng은 ‘지능형 블록 파티션 데이터베이스‘ 구조를 사용하여, 전 세계에 존재하는 미생물 유전체 라이브러리를 굉장히 빠르게 검색합니다.
이를 통해 환자의 장내 미생물 상태를 단 몇 분 만에 진단할 수 있게 되었습니다. [21]
[알기 쉬운 개념] k-mer와 sliding window 동작
유전체 서열은 A, T, G, C로 이루어진 매우 긴 문자열입니다. 이를 통째로 비교하는 것은 속도가 너무 느리기 때문에, 일정한 길이(k)로 자른 부분 문자열인 k-mer를 사용합니다.
- Sliding Window: 서열의 처음부터 끝까지 한 글자씩 옆으로 이동하며 k 길이의 조각을 추출하는 방식입니다.
- 예:
ATGCGTA에서 $k=4$라면,ATGC,TGCG,GCGT,CGTA순으로 추출됩니다.
- 예:
- Minimizer (데이터 축소): 모든 k-mer를 저장하면 메모리가 폭발합니다. 따라서 특정 윈도우 내에서 가장 작은 해시 값을 가진 k-mer 하나(Minimizer)만 선택하여 데이터량을 획기적으로 줄입니다.
- Kun-peng은 35-bp 윈도우에서 31-bp minimizer를 추출하여 중복 계산을 방지하고 정확도를 높입니다.
[알기 쉬운 개념] Kun-peng에서 적용한 고속 데이터 조회의 핵심: IBPDS와 Spaced Seed
Kun-peng이 빠른 이유는 데이터를 찾는 “지도”가 정교하기 때문입니다.
- Spaced Seed Mask (s=7): minimizer 서열 전체를 그대로 해싱하지 않고, 특정 패턴(Mask)을 씌워 중요한 부분만 추출합니다. 이는 염기서열의 미세한 변이(mismatch)에 유연하게 대응하면서도 해시 충돌을 줄여 검색 속도를 높입니다.
- Mask(마스크)를 씌운다는 것의 의미
- 컴퓨터 과학에서 마스크는 특정 비트(또는 염기)를 통과시키거나(1), 무시하는(0) 필터입니다
- 만약 31-bp Minimizer가 있고, 마스크 패턴이
11011이라고 가정해 봅시다 (실제 Kun-peng의 s=7은 훨씬 복잡한 비트 패턴을 가집니다). • Original Minimizer:A T G C G• Mask Pattern:1 1 0 1 1(0인 부분은 무시) • Masked Result:A T _ C G(가운데 G는 해시 계산에서 제외됨) - 즉, 서열 전체를 해싱하는 대신 마스크가 ‘1’인 위치의 염기들만 골라내어 하나의 해시 값(Hash Code)을 만듭니다.
- 어떻게 미세 변이(Mismatch)에 대응하는가?
- 유전체 데이터에는 서열 해독 오류(Sequencing Error)나 개체 간의 변이(SNP)로 인해 단 하나의 염기만 다른 경우가 빈번합니다.
- Contiguous k-mer (일반 방식): 서열이 한 글자만 달라도 해시 값이 완전히 바뀝니다. 즉, 데이터베이스에서 찾을 수 없습니다.
ATGC→ Hash:12345ATAC→ Hash:67890(전혀 다른 데이터로 인식)
- Spaced Seed : 변이가 자주 발생하는 위치를 ‘0(무시)’으로 설정한 마스크를 사용하면, 그 위치가 달라도 동일한 해시 값을 가집니다.
- 만약 마스크가 세 번째 글자를 무시한다면(
1101),ATGC와ATAC는 둘 다AT_C로 처리되어 동일한 해시 값을 생성합니다. - 결과적으로 약간의 오차가 있는 서열도 데이터베이스 내의 동일한 위치(Block)에서 찾아낼 수 있게 됩니다.
- 만약 마스크가 세 번째 글자를 무시한다면(
- Mask(마스크)를 씌운다는 것의 의미
- IBPDS (Index-Based Partitioned Data Structure): 전체 데이터베이스를 하나의 거대한 덩어리가 아닌, 여러 개의 순차적 블록(Sequential Blocks)으로 나눕니다.
- 생성된 해시 코드가 저장 블록 번호와 블록 내 시작 위치를 동시에 결정합니다.
- 덕분에 쿼리 시 메모리 전체를 뒤질 필요 없이 특정 블록만 로드하여 즉시 접근할 수 있습니다.
주목해야 할 생명정보학 도구 목록
| 도구명 | 주 용도 | 기술적 강점 |
|---|---|---|
| MegaPX | 메타프로테옴 분석 | IBF 기반 멀티 인덱싱으로 메모리 효율 극대화 |
| SpliceHarmonization | RNA 스플라이싱 분석 | 치료제 개발을 위한 스플라이싱 이벤트 통합 관리 |
| Tractor Workflow | 국소 조상 기반 GWAS | 다양한 인종 데이터가 섞인 환경에서도 정확한 연관성 분석 |
| scSurv | 싱글셀 기반 생존 분석 | 딥 생성 모델을 활용하여 개별 세포 상태와 환자 수명 연결 |
참고자료
- Tricorder Tech: Protein Sequencing Advance Offers New Insights Into Life’s Foundations, https://astrobiology.com/2026/03/tricorder-tech-protein-sequencing-advance-offers-new-insights-into-lifes-foundations.html
- Volume 42 Issue 3 | Bioinformatics – Oxford Academic, https://academic.oup.com/bioinformatics/issue/42/3
- Volume 27 Issue 2 | Briefings in Bioinformatics – Oxford Academic, https://academic.oup.com/bib/issue/27/2
- Bioinformatics – Oxford Academic, https://academic.oup.com/bioinformatics
- Evolving Strategies for the Incorporation of Bioinformatics Within the Undergraduate Cell Biology Curriculum | CBE—Life Sciences Education, https://www.lifescied.org/doi/10.1187/cbe.03-06-0026
- A data-intelligence-intensive bioinformatics copilot system for large-scale omics research and scientific insights – Oxford Academic, https://academic.oup.com/bib/article/26/4/bbaf312/8196318
- News for News & Commentary – Bioinformatics.org, https://www.bioinformatics.org/news/?group_id=10
- Bioinformatics.org: Home, https://www.bioinformatics.org/
- Volume 26 Issue 4 | Briefings in Bioinformatics – Oxford Academic, https://academic.oup.com/bib/issue/26/4
- Frontiers in Bioinformatics | Articles, https://www.frontiersin.org/journals/bioinformatics/articles
- Call for Proceedings – ISMB 2026 – International Society for Computational Biology, https://www.iscb.org/ismb2026/call-for-submissions/proceedings
- Researchers identify major genetic risk factor for rare form of dementia – Alpha Galileo, https://www.alphagalileo.org/en-gb/Item-Display/ItemId/270149
- (PDF) A repeat expansion in GOLGA8A is a major risk factor for atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions – ResearchGate, https://www.researchgate.net/publication/401959223_A_repeat_expansion_in_GOLGA8A_is_a_major_risk_factor_for_atypical_frontotemporal_lobar_degeneration_with_ubiquitin-positive_inclusions
- Major Risk Factor for Rare Early-Onset Dementia Found …, https://neurosciencenews.com/golga8a-ftd-genetic-risk-30305/
- Rare Dementia Subtype Linked to Newly Identified Genetic Risk – Technology Networks, https://www.technologynetworks.com/neuroscience/news/rare-dementia-subtype-linked-to-newly-identified-genetic-risk-410615
- SHEST: single-cell-level artificial intelligence from haematoxylin and eosin morphology for cell-type prediction and spatial transcriptomics reconstruction | Briefings in Bioinformatics | Oxford Academic, https://academic.oup.com/bib/article/27/1/bbag037/8488669
- Advances and challenges in cell–cell communication inference: a comprehensive review of tools, resources, and future directions | Briefings in Bioinformatics | Oxford Academic, https://academic.oup.com/bib/article/26/3/bbaf280/8169297
- 2026 Most Popular Concentrations in Bioinformatics Degrees – Research.com, https://research.com/advice/most-popular-concentrations-in-bioinformatics-degrees
- Life Science and Biotech Trends for 2026 – AZoLifeSciences, https://www.azolifesciences.com/article/Life-Science-and-Biotech-Trends-for-2026.aspx
- Systematic clustering alignment and feature characterization for single-cell omics using ACE-OF-Clust – bioRxiv, https://www.biorxiv.org/content/10.64898/2026.03.09.710668v1.full.pdf
- Briefings in Bioinformatics | Oxford Academic, https://academic.oup.com/bib
- AI-driven computational methods and benchmarking for T-cell antigen identification | Briefings in Bioinformatics | Oxford Academic, https://academic.oup.com/bib/article-abstract/doi/10.1093/bib/bbag123/8526139
- Closing the loop on HIV-1 circRNAs: Discovery of a functional HIV-encoded circRNA that impacts viral gene expression – Research Communities, https://communities.springernature.com/posts/closing-the-loop-on-hiv-1-circrnas-discovery-of-a-functional-hiv-encoded-circrna-that-impacts-viral-gene-expression
- Advance articles | Bioinformatics – Oxford Academic, https://academic.oup.com/bioinformatics/advance-articles
- scSurv: a deep generative model for single-cell survival analysis – Oxford Academic, https://academic.oup.com/bioinformatics/article/42/1/btaf671/8402136
- The Male Default Prevails in Biomedical Research: Sex Inclusion in Nature (2025) | bioRxiv, https://www.biorxiv.org/content/10.64898/2026.03.03.709344v1
- ECCB 2026 – 25th European Conference on Computational Biology, https://eccb2026.org/
- Call for Papers – BIOINFORMATICS 2026 – SCITEVENTS, https://bioinformatics.scitevents.org/CallForPapers.aspx?y=2014Call
- Reproducible Bioinformatics Analysis Workflows for Detecting IGH Gene Fusions in B-Cell Acute Lymphoblastic Leukaemia Patients – MDPI, https://www.mdpi.com/2072-6694/15/19/4731
- Short Courses in Bioinformatics and Biocomputing (CBRS) | Texas Research, https://research.utexas.edu/cbrs/bioinformatics-short-courses
- Scientific publications 2026 | SIB Swiss Institute of Bioinformatics, https://www.sib.swiss/community/publications/scientific-publications-2026
- Frontiers in Bioinformatics, https://www.frontiersin.org/journals/bioinformatics
- 17th International Conference on Bioinformatics Models, Methods and Algorithms – WikiCFP, http://www.wikicfp.com/cfp/servlet/event.showcfp?copyownerid=90704&eventid=188665
- International Conference on Predictive Analytics in Bioinformatics in San Francisco, USA on 11-March-2026, https://conferencealerts.co.in/event/100237589
- International Conference on Bioinformatics and Biomedicine ICBB on March 16-17, 2026 in Melbourne, Australia, https://conferenceindex.org/event/international-conference-on-bioinformatics-and-biomedicine-icbb-2026-march-melbourne-au
- mnDINO: Accurate and robust segmentation of micronuclei with vision transformer networks, https://www.biorxiv.org/content/10.64898/2026.03.09.710648v1